Page 2 of 8
Part 1: Modelling assumptions and DFT data
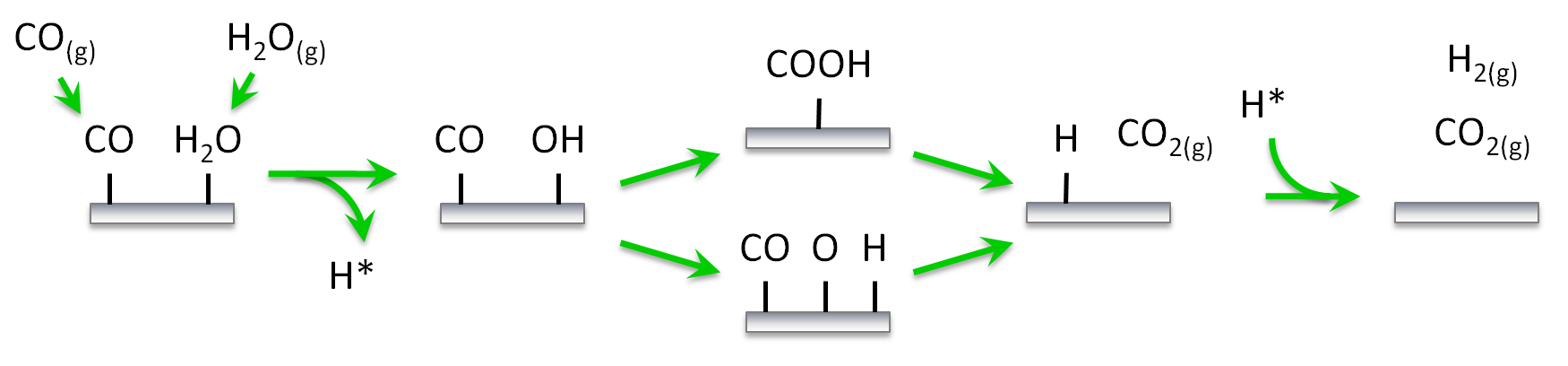
The first pathway we would like to investigate (shown schematically below) involves carboxyl (COOH) as the key intermediate for the water gas shift reaction on Pt(111). The key steps are the adsorption of CO and H2O on the surface, the decomposition of H2O into OH and H, the reaction between CO and OH to form COOH, the decomposition of the latter towards CO2 and H, and the associative desorption of H2. The second pathway includes the dissociation of OH further into O and H and the CO oxidation by O. The data are based on our previous work on water-gas shift,1 although we have made some simplifications for the purposes of this tutorial. Thus, we will assume that: (i) the catalytic surface can be represented by a lattice of equivalent sites (single site type), (ii) dissociative adsorption events are not activated, (iii) lateral interactions are limited to 1st nearest neighbour range and involve only a few key species participating in these pathways, (iv) the unit cell is large enough so that interactions of adsorbates with their periodic images can be ignored. With regard to assumption (i), accounting for more than one site types on catalytic models is straightforward, it may however, require extra care and diligent book-keeping. Assumption (ii) is also made for simplicity in this tutorial, but it is easy to consider transition states for dissociative adsorption events. Assumptions (iii) and (iv) are also oversimplifications for the purposes of this tutorial. Fitting cluster expansions with longer-range interactions, and by taking into account the periodic images will be covered in another tutorial. Moreover, we will not concern ourselves with zero-point energy (ZPE) corrections, these can be added in a straightforward way and everything we discuss applies to the ZPE-corrected energies as well.
The tables below show the energies computed for the species of interest by software package SIESTA (for computational details see Ref. 1).
| Catalytic Surface |
DFT Energy (eV) |
| Pt(111) |
-11448.220 |
| Gas Species |
DFT Energy (eV) |
| CO |
-588.789 |
| H2O |
-467.460 |
| H2 |
-31.403 |
| CO2 |
-1025.462 |
| O2 |
-867.202 |
| Surface Species |
DFT Energy (eV) |
| CO* |
-12039.087 |
| H2O* |
-11916.042 |
| OH* |
-11899.149 |
| O* |
-11882.980 |
| H* |
-11464.540 |
| COOH* |
-12490.256 |
| Co-Adsorbed Species (1NN) |
DFT Energy (eV) |
| CO*+CO* |
-12629.394 |
| OH*+H* |
-11915.448 |
| O*+H* |
-11899.102 |
| CO*+OH* |
-12489.950 |
| CO*+O* |
-12473.424 |
| Transition States |
DFT Energy (eV) |
| TS1: H2O*→OH*+H* |
-11915.265 |
| TS2: OH*→O*+H* |
-11898.209 |
| TS3: CO*+OH*→COOH* |
-12489.544 |
| TS4: COOH*→CO2+H* |
-12489.404 |
| TS5: CO*+O*→CO2 |
-12472.435 |
In the following section we will discuss how to choose a set of reference species with respect to which the formation energies of all species will be calculated.