A simple KMC simulation
There is a lot of theory and mathematical formalism behind the kinetic Monte Carlo (KMC) algorithm and how/why it works, but for all practical purposes it can be thought of as a computer game that uses random numbers. The game is being played on a lattice, each site of which can be occupied by either of the two adsorbates of our example, CO* or O*. The KMC algorithm performs random "moves" (referred to as elementary events) e.g. adding a CO* adsorbate via adsorption at a certain lattice site at some time t1, performing a diffusional hop at some time t2, adding a pair of O* adsorbates via dissociative adsorption at neighbouring sites at t3, performing a diffusional hops of a O* at a time t4 etc. When O* and CO* "meet each other", they also have a certain probability of reacting towards CO2 gas, so KMC also simulates such events once in a while. Important points to note are that a typical KMC algorithm:
- executes/realizes elementary events one after the other (one event at time)
- uses random numbers to decide when the next event will take place (t1, t2, ...) and what kind of event it will be (adsorption, desorption, diffusion, reaction etc.)
- generates these random numbers in such a way that events with higher kinetic constants are executed more frequently, as expected (i.e. these random numbers are not "completely random").
In fact, there are theorems from mathematical physics assuring us that if we simulate a KMC model for CO oxidation which is a faithful representation of Langmuir-Hinshelwood kinetics, we should get the exact same predictions as the equations listed in the previous section. This is shown in the simulations below:
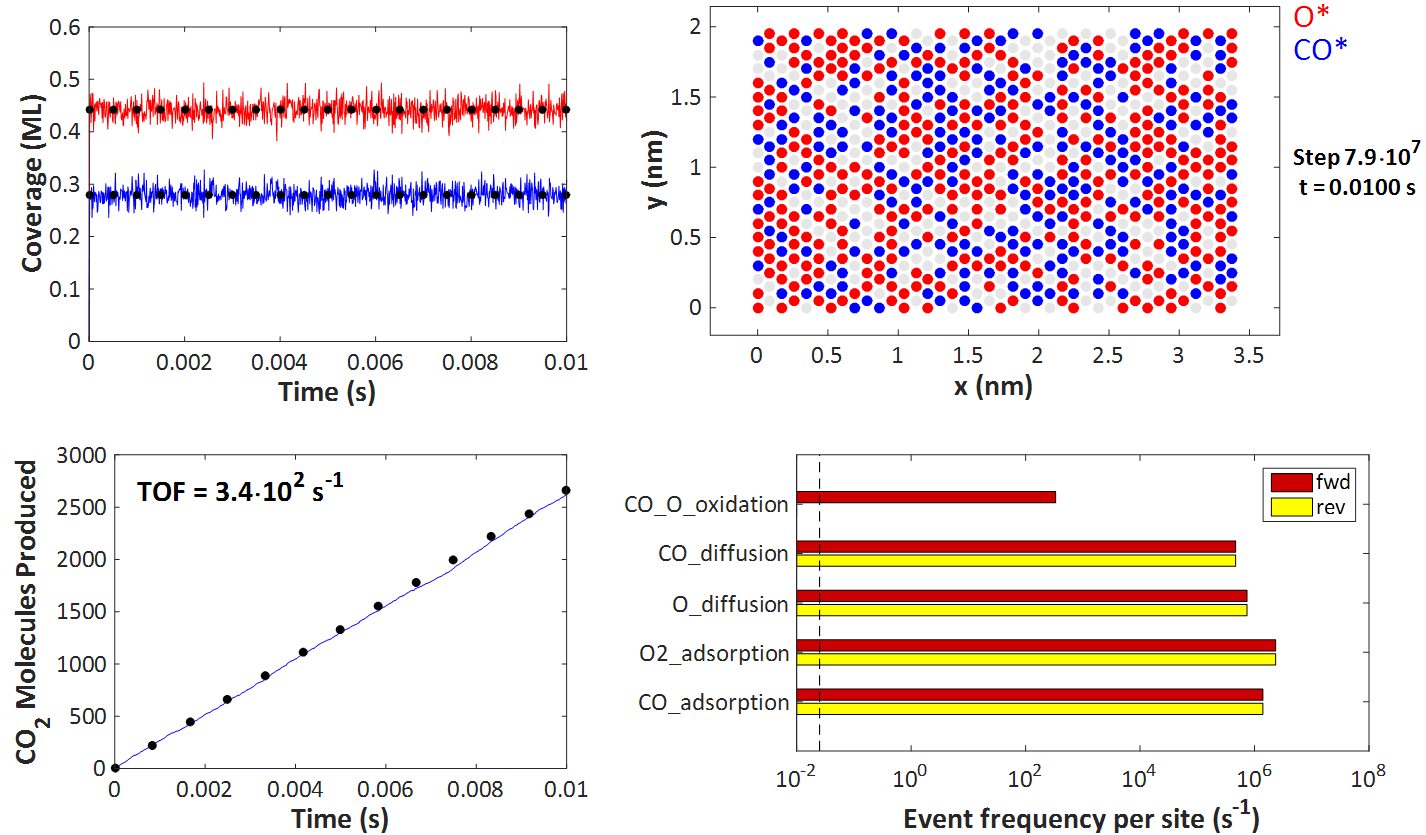
The top left plot depicts the coverages of O* and CO* over time, which quickly reach steady state (the transients are too short to be apparent). The black dots represent the predictions of the Langmuir-Hinshelwood model, and it is evident that KMC agrees with this model. Of course, there is some noise because of the random (stochastic) nature of KMC, but the aforementioned theorems inform us that simulating larger and larger lattices will bring the noise down, making the KMC predictions indistinguishable from those of the Langmuir-Hinshelwood deterministic equations.
On the top right plot, one can see a snapshot of the KMC lattice at the end of the simulation. The filled red circles correspond to O*-occupied sites, the blue ones to CO*-occupied sites, whereas the grey circles denote vacant sites. As per assumptions 1 and 2 of the Langmuir-Hinshelwood model, discussed earlier, each site can only be occupied by at most one adsorbate molecule, and all sites are equivalent. We also have no lateral interactions in the KMC model, so there is no discernible ordering on the adlayer.
The bottom right figure compares the KMC prediction for the number of CO2 molecules produced over time (blue line) with that of the Langmuir-Hinshelwood model. In particular, the black dots are on a line with zero intercept and slope equal to RateLH from the equations above. Again, the KMC prediction closely matches that of the Langmuir-Hinshelwood model.
Finally, the bottom right plot portrays a histogram of the reaction statistics. Worth noting is that the event frequency of the CO_O_oxidation event is the turnover frequency (TOF) of 340 s-1, in agreement with the plot on the left just discussed. Moreover, adsorption/desorption and diffusion events are quasi-equilibrated and occur on a much faster timescale than the oxidation event. These observations are in line with assumptions 3 and 4 of the Langmuir-Hinshelwood model (note also that the KMC kinetic constants are indeed constants, i.e. independent of coverage in this simple example).
If you wish to run these calculations in Zacros and check your results, you can obtain the full input and output files via the link below:
Download LangmuirHinshelwood.zip.
A technical point to note when reproducing these results: the value of koxi in the Langmuir-Hinshelwood model must be 6 times the value of the rate constant for the CO_O_oxidation event fed as input to the KMC. The reason is that in our lattice, each site has 6 neighbours (coordination number 6), so the oxidation event is "replicated" across each neighbouring site. Ref. 1 contains more information on the mathematics of this.