Part 4: Reaction mechanism
The reaction mechanism is described in the mechanism_input.dat file. The file starts with the keyword mechanism, and ends with end_mechanism. The contents of the file consist of blocks starting with keyword step (for irreversible elementary steps) or reversible_step (for reversible elementary steps) and ending with end_step, or end_reversible_step, respectively. It is generally recommended to use only reversible steps in KMC simulations otherwise the simulation may violate detailed balance and thermodynamic consistency. For this tutorial we are implementing the ZGB model which is a prototypical irreversible model, so we will only use step … end_step blocks.
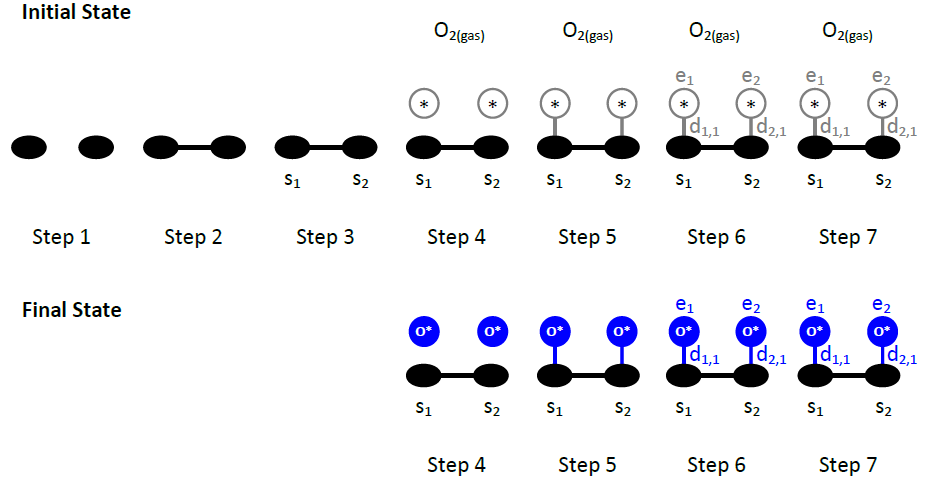
The way one defines the initial and final states of the elementary steps is very similar to the definition of the lattice state in energetic patterns. Thus, to correctly define elementary steps, one can draw pictures of the initial and final states following steps similar to 1-7 outlined in the previous section:
- Draw all the sites involved in the initial state of the elementary event.
- If more than one sites are involved, draw all the links between the sites indicating which sites are neighbours.
- Number all the sites in the pattern, as s1, s2,…
- Draw all the gas species and adsorbate entities participating on the pattern, clearly indicating the species type for each entity. An empty site is assumed to be a site occupied by a “vacancy” pseudo-species.
- Draw the bonds between adsorbates and sites, to clearly indicate which adsorbate occupies which site (or sites, if there are multidentate adsorbates).
- Number all adsorbates (entities) as e1, e2,…
- Number all dentates as d1,1, d1,2, …, d2,1, d2,2,… The first index in this numbering system is the entity number, and the second is the dentate number.
- Repeat steps 4-7 for the final state of the pattern, using the numbering of the sites consistently in the initial and final states of the pattern.
As an example we will demonstrate these pictures for the O2 dissociative adsorption event:
The Zacros instructions for this patter are as follows:
step O2_adsorption
for the definition of the name of the event.
The following line defines the gas phase reactants or products: the keyword gas_reacs_prods can be followed by an arbitrary number of pairs of a gas species name and a stoichiometric coefficient. The latter can be negative, defining a gas phase reactant, or positive defining a product. In our case, an O2 gas molecule is removed upon adsorption.
gas_reacs_prods O2 -1
The next two lines indicate that there are 2 sites involved in this event, which have to be nearest neighbours:
sites 2
neighboring 1-2
In the initial state, we have two empty sites on the lattice. Thus, both sites are “occupied” by the vacant species, denoted by the reserved name *. The description of this initial state consists of the keyword initial, followed by 2 lines of 3 words, the 1st line corresponding to site 1 and the 2nd to site 2:
initial
1 * 1
2 * 1
Note that we are consistent with the numbering of the figure above: the first (second) site is occupied by the first (second) entity. The entity numbering makes each line unique in a state specification. Having one or more lines with the exact same information within a state specification (either initial or final) is invalid and will result in an error.
In the final configuration, there are two O adatoms. Consistent with the figure above, we write:
final
1 O* 1
2 O* 1
Further, we have to define a pre-exponential and an activation energy for this event:
pre_expon 2.5
activ_eng 0.0
With an activation energy of 0 eV, the kinetic constant of this event is always equal to the pre-exponential value.
Finally, the block ends with the end_step keyword.
The blocks for the CO adsorption and the CO oxidation steps are shown below, followed by the Zacros input for these two events:
step CO_adsorption
gas_reacs_prods CO -1
sites 1
initial
1 * 1
final
1 CO* 1
pre_expon 10.0
activ_eng 0.0
end_step
step CO_oxidation
gas_reacs_prods CO2 1
sites 2
neighboring 1-2
initial
1 CO* 1
2 O* 1
final
1 * 1
2 * 1
pre_expon 1.e+20
activ_eng 0.0
end_step
With regard to the rate parameters for the elementary events, since the original ZGB implementation uses a null-event type of approach, we need to properly choose our kinetic constants to simulate the same behaviour. Thus, the CO oxidation event rate was taken as very high, since in the original ZGB model this event happens immediately when a CO-O pair is formed. The CO adsorption rate pre-exponential was arbitrarily taken as 10 bar-1s-1. Thus, if the partial pressure of CO in the gas phase is 0.45 (see part 1: Simulation setup) the rate will be 4.5 s-1. The O2 adsorption pre-exponential was chosen to be ¼ that of CO: this way, because of the coordination number 4 of the lattice, for every empty site therein, the maximum O2 adsorption rate can be 10 bar-1s-1, when all neighbouring sites are vacant. This setup mimics the null-event approach by Ziff, Gulari and Barshad, wherein for equimolar CO:O2 mixture, an empty lattice has equal probability of being picked for a CO or O2 adsorption event, and the success of an O2 adsorption trial is subsequently determined by looking at the neighbouring sites.