Part 2: Input for single-body terms
Preliminary schematics in preparation for input
We now proceed to set up the Zacros energetics_input.dat file describing our cluster expansion. To correctly define the patterns in energetics_input.dat, it is often helpful to make a schematic of the binding configuration wherein we clearly number the sites, adsorbate molecules and dentates thereof. This drawing process can be accomplished by following these steps:
- Draw all the sites involved in the pattern.
- If more than one sites are involved, draw all the links between the sites indicating which sites are neighbours.
- Number all the sites in the pattern, as s1, s2,…
- Draw all the adsorbate entities participating on the pattern, clearly indicating the species type for each entity.
- Draw the bonds between adsorbates and sites, to clearly indicate which adsorbate occupies which site (or sites, if there are multidentate adsorbates). At this stage, sites with unspecified states will be numbered but not occupied by any adsorbates. If desired, mark these sites explicitly as unspecified.
- Number all adsorbates (entities) as e1, e2,…
- Number all dentates as d1,1, d1,2, …, d2,1, d2,2,… The first index in this numbering system is the entity number, and the second is the dentate number.
Let us first demonstrate these steps for two very simple patterns: the "AnySite" pattern that contributes the background energy of 12 meV per site irrespective of state (vacant/occupied), and the "O_Point" pattern for a single oxygen adatom on an fcc site.
The figure below shows the steps for drawing the "AnySite" pattern: notice that there is no change from step 1 to step 2, since there is only one site involved (no neighbouring structure). Similarly, there is no change from step 3 to 4, since there a no adsorbates participating in this pattern. The state of the site is unspecified, as indicated by the open marker in step 5. Subsequent steps are not performed, since there are no adsorbates to assign numbers to.

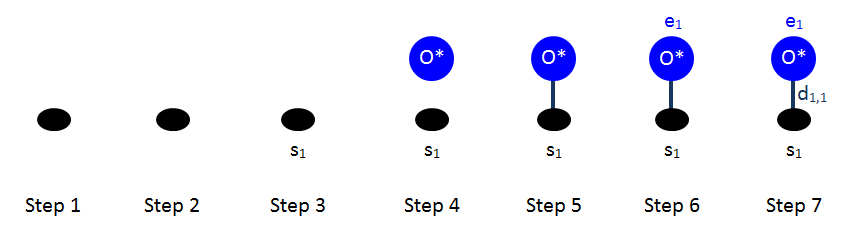
For the case of the oxygen adatom, the drawings are shown below. In this case, we have one adsorbate bound with dentate number 1 (its only dentate) to the single site of the pattern.
Input for the single-body terms
We will now work on the Zacros input that defines these patterns. The file energetics_input.dat always starts with the keyword energetics, followed by one or more blocks of instructions contained within keywords cluster … end_cluster. Each such block describes an energetic pattern. The parsing of the file finished when Zacros encounters the keyword end_energetics.
Thus, for the "AnySite" pattern we write:
cluster AnySite
Then, the number of sites involved (one) is defined as follows:
sites 1
Subsequently, we define the occupancy of the site involved, which is done with the lattice_state keyword. This keyword is followed by as many lines of input as the number of sites in the pattern (in this case only one). Each line contains three words: the first is the number of the entity/adsorbate occupying the corresponding site, the second is the species name occupying that site, and the last word is the dentate number with which the entity binds to that site. In the case of a site with unspecified state, all three words are replaced with the character &:
lattice_state
& & &
Subsequently, we can optionally define the types of sites and the graph multiplicity of the pattern, and we have to specify the pattern's ECI (the energy contribution). The site types are defined by the optional keyword site_types, followed by as many words as the number of sites in the pattern. These words have to correspond to names of site type as defined in file lattice_input.dat. In our case, we assume that we work with a single site type named "fcc_t". We can thus mention explicitly that we require the sites to be of type "fcc_t" for this pattern to be detected:
site_types fcc_t
This specification is however not compulsory: if we don't mention anything about the site types, Zacros will detect the energetic interaction patterns on the lattice based on the neighbouring (connectivity) of the sites and their state. Here we have a single site type anyway, so including or omitting the site_types keyword makes no difference.
The multiplicity is defined by the optional keyword graph_multiplicity, whereas the energy contribution is specified with the keyword cluster_eng. If graph_multiplicity is absent, a default value of 1 is used for the multiplicity. This feature exists for cross-compatibility with other KMC software codes: when Zacros detects this pattern on the lattice, it will add a contribution to the total energy equal to the cluster energy divided by the multiplicity. This is useful, for instance, in the case of a symmetric pairwise pattern, which will be detected twice by the Zacros algorithm. A multiplicity of 2 will correct for this double-counting, as will be discussed in the next section (Part 3). In our case, we have a single-site pattern so the multiplicity can only have a value of 1. Moreover, the ECI value is written in units of eV:
graph_multiplicity 1
cluster_eng 0.012
The cluster specification block ends with the keyword end_cluster.
For the case of the oxygen adatom, we have to specify the state of the site involved in the pattern. We thus observe that site number 1 is occupied by dentate 1 of entity number 1, which is a O* (see figure above). The site type is fcc_t, the graph multiplicity is 1, and the cluster energy is -1.368 eV. Therefore:
cluster O_Point
sites 1
lattice_state
1 O* 1
site_types fcc_t
graph_multiplicity 1
cluster_eng -1.368
end_cluster